Particle operations
Access the particle operations menu by clicking on a species’ particle count.
Intersect with another particle set
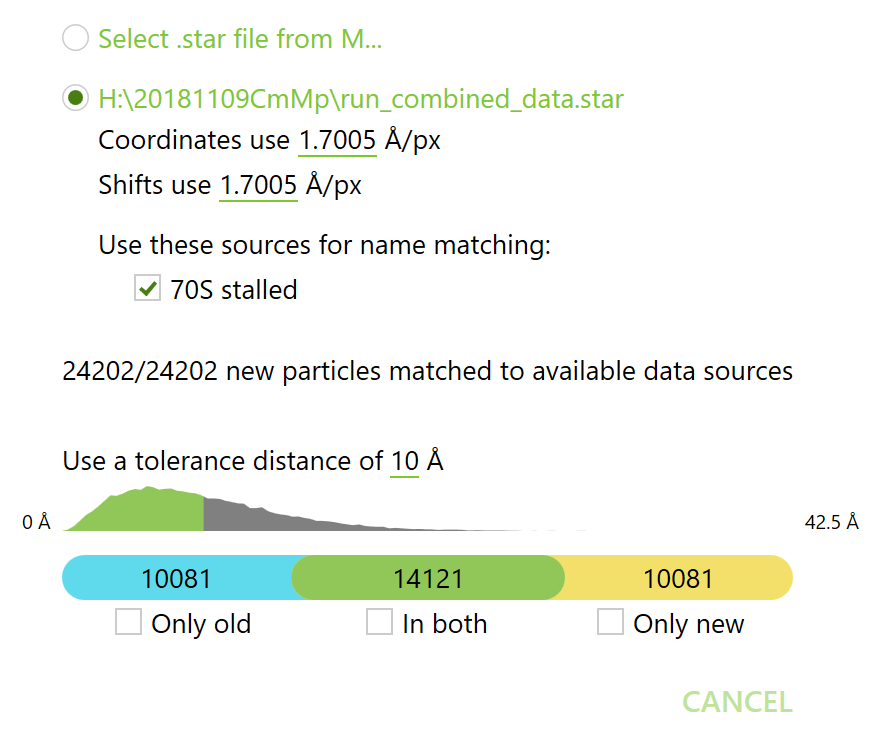
This dialog allows you to intersect the current particle set of a species (“old set”) with another particle set defined in a STAR file (“new set”).
When loading particles from another M species, the source frame/tilt series are matched unambiguously, and the coordinates are always in Ångstrom. When loading a refinement or classification result from RELION, the data sources must be matched manually in case of name duplications, and the pixel size for the coordinates must be provided.
Once loaded, particles matches between the two sets are found by solving the unbalanced assignment problem with the inter-particle distance as the cost function. You can specify a maximum tolerance distance for the matching, beyond which any matches will be dismissed. The inter-particle distance histogram can help you with that.
The matching creates 3 sets: “only old”, “in both” (intersection), and “only new”. The particles in the intersection will keep their old poses. You can select any combination of them and update the species’ particle list with the resulting set. For instance, if you want to add completely new particles (e. g. for a data source you didn’t use before), set the tolerance distance to 0, and select “only old” and “only new”. If you want to remove certain particles, set the tolerance distance accordingly and select “only old”. If you want to keep only particles remaining after another classification run, select “in both”. To remove particles that shifted too much during M’s refinement, select the STAR file from the first version of the species, set the tolerance distance, and select “in both”. The possibilities are quite limitless, yes, yes!
Export sub-tomograms

M can refine per-particle pose trajectories. To consider them for export, please use this dialog rather than Warp’s particle export. Some field values will be pre-populated based on the species’ parameters, but feel free to change them. Particle export from frame series is currently not supported.
Output will be scaled to…: The target pixel size to which the tilt series data will be scaled prior to reconstruction. It can’t be smaller than the size dictated by the Bin parameter in the Input settings section. The maximum resolution achievable with the data will be twice the pixel size.
Box size is…: The sub-tomogram box size in pixels. M avoids CTF aliasing by calculating the necessary minimum box size for the particle’s defocus value and the expected maximum resolution, as dictated by the pixel size, and then crops the volume back to the value you specified. For high-defocus data and small boxes, this can make the high-resolution Thon rings invisible. Don’t worry, this is fine. In fact, it is much finer than previous methods that would have made the data useless beyond the aliasing-free resolution.
Particle diameter is…: The particle diameter should be roughly the same you will use later in RELION, otherwise it might complain about incorrect normalization.
Volumes / Image series: In addition to reconstructing 3D sub-tomograms, M can also generate a series of 2D tilt images for each particle. There is currently no use for this data type in RELION, unless you have giant particles with a lot of signal and would like to refine the particle tilt images completely independently like in EMAN 2.3. This is probably not a good idea.
Shift by…: You can shift all particle positions by the same amount in 3D. If the input STAR file contains refined particle orientations (rlnAngleRot/Tilt/Psi columns), the shift will be made in the refined map’s reference frame. Use this if you have aligned all particles on a common part of the protein, and want to e. g. shift the map center to a ligand to analyze it further. Note that the shift values are in Angstrom.
Pre-rotate particles: If refined particle orientations (rlnAngleRot/Tilt/Psi columns) are available, the reconstructions can be pre-rotated to their common reference frame. If you were to average the resulting sub-tomograms without considering any rotations (e. g. simply sum them up in Matlab), you would get the refined map. This can be useful if you’re developing a new method to analyze the particles (e. g. variance, 3D PCA etc.) and don’t want to bother with particle orientations. The 3D CTFs will be pre-rotated to the same reference frame, too.
Limit to first N tilts by dose: The reconstructions or particle tilt series will contain only data from the first N tilts, sorted by dose. This can be useful if you think the late tilts don’t contain any information that might help your analysis.
Invert contrast: Multiply the input data by -1 to invert the contrast. Following standard conventions, black particles from cryo data will become white if this is enabled.
Normalize input images: The scaled tilt images will be normalized to mean = 0, standard deviation = 1 prior to reconstruction.
Normalize output volumes: Normalizes the reconstructions so that the volume outside the specified particle diameter is mean = 0, standard deviation = 1. This is required for RELION.
Make 3D CTF sparse: Don’t use this just yet! It requires a patch for RELION that isn’t public yet. If enabled, every voxel of the 3D CTF with a value below 0.01 will be explicitly set to 0. This allows the patched RELION algorithms to completely exclude them from calculations, saving a lot of time.
After clicking Export, M will ask you to specify the name and location of the output STAR file that will contain the paths of all volumes and their metadata. Use this file as input for RELION.